|
BIOL
4160
Evolution
Phil Ganter
301 Harned Hall
963-5782 |
Looking
up at a Redwood (Sequoia sempervirens) |
Systematics
Email me
Link
to a list of Specific
Objectives for lectures
Back
to:
Classification is the process of subdividing large collections of items,
living or not, into identifiable groups based on a rule or set of rules
- The need for this comes out of the power of organization to allow a person
to think about large groups of things
- Libraries must classify their holdings
so that an individual can find a particular item without a piece-by-piece
search
- Biological Classification arises from the same need because there are
so many different kinds of organisms -- too many to think about without
resorting
to grouping them
Taxonomy
- Any system of organizing things based on shared characteristics is a
taxonomy
- taxonomy does not have to reflect common ancestry, only similarity
- One can construct a taxonomy of buttons
based on size, number of holes, materials used, shape, etc. but this
taxonomy would not reflect
anything
about the "ancestry" of buttons
Systematics is
the classification of biological diversity through the use of shared
ancestry (relatedness)
Biological Evolution is not merely
change in organisms
Groups of related organisms
evolve, not individual organisms nor, as the book says, simply groups
of organisms
Relatedness and Inheritance
Putting aside the question of the origin of
life, we assume that all new organisms are the outcome of reproduction
by organisms in the previous generation. That is, all new organisms
have a parent or parents.
- This assumption gives rise to the concept
of relatedness - connections
between organisms due to the material inheritance offspring receive from
their parent or parents.
- In biological evolution, inheritance
is material but it's really more complicated than that
- The matter (DNA and other information-rich
molecules in the gametes) inherited carries information about
the structure of the organism and structure is related to
function
- Because we can
consider information as inherited, then
all information
received from other members of the
same species can also be considered an
organism's inheritance
- Social species then receive
both genetic information and cultural information
- A corollary of the idea of relatedness is
the idea of distance in relations. This arises because an organism's
parent or parents have, in turn, their own parent or parents. Relatedness
connects organisms across many generations.
Darwin (and others before him) saw all life
as descending from a single origin. In this view of life, all
living organisms are related, although the distance between some
has grown great because the common parents they share are many, many generations
in the past.
- Why base biological classification on shared ancestry
- Biologists desire a natural system of classification
- Natural systems
are those whose existence does not depend on the presence of
humans - we discover these systems but we do not
invent
them
- Artificial systems are those that do depend on our presence -
we invent these systems but we can not discover them
- Classification can be either natural or
artificial but natural classifications tell us more about the biological
world than do artificial ones
- A Darwinian view of evolution, whether by natural
selection or not, involves the application of the idea of ancestry (originally
used to describe the relation of parent to offspring) to groups of organisms
- from populations to species and larger groups
- Just as a genealogy branches out over
time from a single individual, relationships between groups of organism
branch out
from a Common Ancestor
- For any three or more groups of organisms, the
two with the most recent common ancestor are the most closely related
- Primitive - occurring or originating long ago
- Derived - occurring or originating more recently
Phylogenetics is the study of relationships among
groups of organisms based on relatedness (common ancestry)
- Phylogenetics can also be seen as the study of
the evolutionary history of organisms, assuming the Darwinian view of evolution
Inferring Phylogenetic
History
We will never know with certainty any phylogenetic
history prior to today
- No one recorded the data
- So, we must infer the history from the existing
data
The first systemetists had several kinds of
data: morphology,
anatomy, behavior, habitat
- They made an assumption in order to derive an
evolutionary history from the data:
- Organisms that are closely related are more likely
to share a trait than are less closely related organisms
- Many recognized the faults in the assumption
of similarity = relation
Phylogenies are based on ancestry but they
are still constructed on the assumption that degree of similarity directly
indicated degree of relatedness
-
Some definitions
- Taxon (Taxa pl.) is a group of organisms
classified into a single group -
- a phylogenetic tree normally
has all terminal taxa with similar taxonomic rank (all
species or all populations within a species,
etc.) although many trees
drawn to illustrate particular points violate this
- Characters (Traits)
are features of an organism
- Characters may take on values (Character States)
and these may be:
- Continuous - the character
state may be one of an infinite set of states within the total range
- Discrete - character states can have only certain
values within the total range of values
- Ancestral states are those present in the ancestor
of any set of taxa
- Derived
states are those character states
found only in a subset of the descendents
of a single ancestral taxon
- The ancestral
state is an Plesiomorphy (pronounced
Please-e-o-morphy) and the descendent
(=derived) state is an Apomorphy
- Terminal Taxa are those taxa at the tips of a tree's branches (that
have no descendent taxa) and, for most trees, they are the living taxa
- Other taxa all are made of an ancestral taxon and its descendents
and there are three types:
- Monophyletic - a taxon is monophyletic if it includes an ancestral taxon
and all of
the
taxa
that are
descendents of the thet ancestral taxon
- Paraphyletic - a taxon is paraphyletic
if it includes an ancestral taxon and some, but not all, of its descendent
taxa
- Polyphyletic - a taxon is polyphyletic if it
includes an ancestral taxon and at least one other taxon that is
not a descendant of the ancestral taxon
- Hennig formalized these ideas about similarity
and relatedness when he proposed that there are three reasons for two
organisms to share a character state
- Ancestral Inheritance -
the character state was in the ancestral taxon
- Therefore, it should be in all of the ancestral
taxon's descendents
- If it is missing, it has been lost or altered
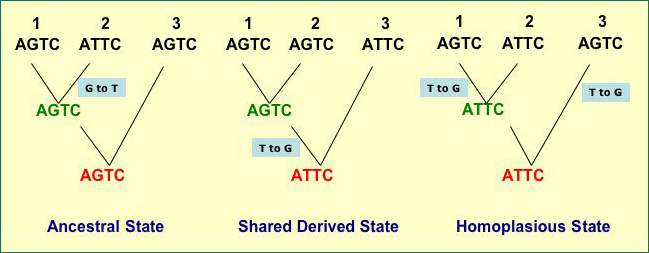
- The tree on the left in the figure below illustrates
this
- Taxa 1 and 3 share a guanine in the second
position in the short DNA sequence that is not shared by taxon 2
- They do so because the ancestor of the group
(the sequence in red) has a G
- Inheritance of Shared
Derived Character States (also called Synapomorphies)
- the character state was not present in the ancestor for the
entire
lineage
but is
found in two or more taxa because they chare a recent ancestor
with that trait
- The tree in the middle in the figure below
illustrates this
- Taxa 1 and 2 share a guanine in the second
position in the short DNA sequence that is not shared by taxon 3, as
on the tree to the left
- They do so because their most recent ancestor
has that state (G) but the ancestor did not
- The state of "G" is
derived because it is no- t found in the more distant ancestor
but is found in a more recent
ancestor
- Thus, the "G" is a shared,
derived character
- For the sake of completeness,
I should mention that an apomorphy that is not shared (i.
e. it is found only in one taxon) is called an Autapomorphy and is useless when constructing a phylogeny
- Homoplasy
- Two or more taxa share a state because
the state arose more than
once. There are two reasons
for this
- Convergence - the state arose in different
lineages within the tree
- Reversal - the state arose twice in
the same lineage
- Analogy is
the older term and was used to describe convergence of phenotypic
character states
in unrelated lineages (like the similarities [thorns vs. spines, storage of water
in stems, loss of leaves] between cacti and some euphorbs from
the desert regions of southern Africa)
- Reversals are very rare for complex
anatomical or morphological characters, so analogy was the
appropriate term when molecular data was not available
- Sequence data contains more reversals
(e. g. A mutates to T and back to A again) and homoplasy
is now the more acceptable term
- The trees in the figure
below illustrates why plesiomorphy and homoplasy can mislead
a systemitist (assume the tree branching is the true history
of taxa 1, 2, and 3)
- Left hand tree - Taxa 1 and 3
share a guanine in the second position in the short DNA sequence
that is
not shared by taxon 2
but do so because their ancestor had G there, so the similarity between 1 and
3 based on the second site is due to Symplesiomorphy
- Left Hand Tree - Once again, Taxa
1 and 3 share a guanine in the second position. They
do so not because their most recent ancestor has that state
(the
ancestor
in
red is
their
most recent common
ancestor and it has) but because the state has arisen
twice through mutation and the similarity is due to Homoplasy
- Middle Tree - Now it is Taxa 1 and
2 that share the guanine. The do so because of a mutation
that arose during the period of time depicted by the tree,
so the guanine is a Symapomorphy and is a reliable indicator
of relatedness
- Hennig drew the logical conclusion that, of
the three reasons for shared states, only shared, derived states
(synapomorphies) give any information about relatedness
Constructing a Phylogenetic
Tree
- Given a set of taxa and the character states
of multiple characters, the problem is to draw a tree which reflects the
phylogeny of the group
- Phylogenetic Since we have seen that the
only information useful in this is that found in shared, derived
character
states, we need to separate them from changes that reflect homoplasy
or similarities due
to shared, ancestral states
- Problem 1 - which state of a character is ancestral and which
is derived?
- Problem 2 - has a derived state arisen more than once in a tree?
- If you know what the ancestors' character states
were, this would be a snap but it is exceedingly rare to know this
- Fossils may provide the data that orders the character states
from oldest to newest
- If a closely related group or taxon is included in the analysis,
then this Outgroup can supply evidence about
the order of states (use of an outgroup ROOTS a tree in phylogenetic
jargon)
- Several methodologies have been developed
to construct trees from datasets (we will mention only four)
- Distance methods (also called similarity methods)
- a formula is applied to the data to calculate
the distances (or similarities) among all of the taxa and a procedure
(there are many) is used to take
the matrix
of distances (or similarities) an construct the tree
- Maximum Parsimony - here trees are compared directly
- Each tree is given a "length" -
the total number of evolutionary events that have to occur for
the data to fit the tree
- The tree with the least number of events is most
probably shows the true relationships
- Parsimony is the empirical principle that the
explanation with the fewest number of assumptions is most likely to be
correct
- In this case, each evolutionary event needed
to fit the data onto a tree is an assumption about the evolutionary history
of the taxa, so the tree with the fewest number of events is most parsimonious
- Maximum Likelihood - these methods use a model
of evolution to calculate the likelihood of the data given a particular
tree
- The tree with the largest likelihood is the one
that reflects the true relationships among the taxa
- The evolutionary model is crucial - if it is
wrong, then a false tree may have the greatest likelihood
- Bayesian Probability - this method is based on
Bayes Theorem and uses a model of evolution to compute the probability
of a particular tree given the data
- The most probable tree is the best guess of the
real relationships, given the data
- the model of evolution must be explicit about
the chance of particular mutations occurring (only models of sequence evolution
are explicit enough to be used in this approach)
- Performance
- Various tests have been devised to test the
performance of these methods
- Many involve simulated data (so that
the true tree is known)
- If applied correctly (so that their
assumptions are met, a big if in many cases), then
- Distance Methods are very good and require
the fewest number of calculations
- Maximum Parsimony is excellent but takes more
effort than distance methods
- Maximum Likelihood methods are
better than parsiminoy or distance metods and require even
more effort
- Bayesian Probability is as good as maximum
likelihood and is more efficient (and, although last to be developed,
is becoming the standard)
- Distance methods differ in a very important
way from the other three
- Although there is more than one
method for drawing a tree from a matrix of distances, each
method yields
only one tree (there
are some minor exceptions to this rule, primarily when one
or more branches has a distance of zero)
- The other methods must be applied to every possible
tree that can be drawn from the set of taxa and the best tree is known
only after all of them have been evaluated
- This rapidly becomes a Herculean task
- Given a
set of taxa, the number of rooted or unrooted trees that can be drawn
that
connects
all
taxa becomes
astronomically
large as the number of taxa goes up (more rooted than unrooted
trees for the same number of taxa)
- for 20 taxa, the number of rooted trees is 8.2
x 1021
or 8 thousand trillion trillion trees
- If the fastest supercomputer could examine a
tree in the time it takes it to perform one calculation (called a FLOP),
it would take a year to examine all of the trees (in actuality, thousands
of calculations are needed to evaluate a tree of that size, so a the fastest
computer would take thousands of years!!)
- for 57 taxa, the number of rooted trees is 3.85
x 1090 trees,
or about 4 trillion trillion trillion trillion trillion trillion trillion
trillion trillion trillion trees
- there are only about 1 x 1089 protons
in the universe
- Heruistic Search
- Thus, we cannot really compare
all trees when the number of taxa gets beyond the teens
- The accepted approach is to
search for the best tree without trying out all of them
(we haven't time to discuss how this
is done) - called a heuristic search
- The performance evaluation
above is based on heuristic methods
Molecular Clocks
A molecular clock is the ability to measure time
by measuring change to a DNA or protein sequence
- By comparing orthologous sequences in two taxa,
we could then tell how long ago they shared a common ancestor
- Requires several assumptions
- constant rate of change all along the sequence
and between the two lineages
- no homoplasy (reversals, etc.)
- If we know the rate of single changes, we can
get an absolute time since their common ancestor
- A tree with tree branch lengths that reflects
the distances between taxa presents a view of the relative
time since divergence but absolute time since divergence is possible
if the clock can be calibrated
- Two ways to calibrate the clock
- fossil data on a common ancestor
- measuring the rate of neutral change in the sequence
- Molecular clocks are popular but still controversial
- It is known that some lineages violate the constant rate assumption
- Relative Rates Test
- Since the amount of time that has elapsed
since any two taxa shared a common ancestor is the same for both
taxa (no matter how many splits into new taxa have occurred during
that time), the number of changes should be the same for both taxa
with random chance explaining any differences found
- Relative Rate Test tests the assumption
of equal number of changes in two lineages (first a tree must be
constructed and the number of evolutionary events counted on the
tree)
- For closely related species, the relative
rate test often finds no difference
- For distantly related species, the relative
rate test finds many more cases of different rates of evolution in
the two lineages compared
- This is a direct test of the most important
assumption behind molecular clocks: constant rates of evolution
Phylogenetic Problems
There are recognized problems in tree construction,
some are theoretical and some are practical
- Incongruence
- Early in the "Sequencing Era",
a single gene was sequenced in several species or populations and a
phylogeny of the species,
not the gene, was inferred from the data
- This assumes that all genes in an organism
have the same evolutionary history, so all loci reflect the history
of the species
- However, as sequencing became easier and more common,
multiple genes were often sequenced and combined in a single phylogeny
- This is done in two ways:
- All data is combined into a single analysis
- A tree is constructed for each gene sequence and
the species tree is the consensus among the different gene trees
- Method 1 forces a consensus from the data by assuming
that all gene histories are those of the species and deviations are due to
undetected homoplasy or simply error in data collection
- Method 2 does not make the same
assumption and has discovered that genes in the same individual
may have different
ancestries
due to such evolutionary events as horizontal transfer
of genes, hybridization, gene
duplication, and confusion due to
polymorphic
characters inherited by
descendents (this confusion is generally said to be caused
by "Lineage
Sorting")
- Lineage sorting is the result
of the presence of polymorphic loci that persist
over one or more speciation events.
- Suppose
Species A splits into Species B and C
and Species C further splits in to Species
D and F.
- B, D, and
F are the extant species and you collect
data on the
same gene from all three.
- Locus
W (for fuzziness, say) is polymorphic
in ancestral species A (W1W2,
W1 produces
fuzz, W2 does
not)
- The polymorphism
persists in ancestral species
C
- Over time, W1 become
fixed in species B
- When Species C splits into Species D and
F, W2 becomes
fixed in Species D, but W1 is
fixed in Species F
- Your
data now shows that Species B and
F share allelel W1 and
Species D has allele W2,
even though the true tree shows
that species D is most closely
related to species F, not species
B
- Notice that this problem
arose without
any convergent evolution or mutation and can arise
from any locus that
is polymorphic at the time of speciation
- The use of method 2 has, in
fact, been key to uncovering instances of horizontal
gene transfer (see below) and hybrid species formation
(see below)
- Problems with Character Scoring
- Phenotypic characters
- How to score multiple changes (and
even deciding how many changes actually took place!) can be
difficult and, if you don't get it right the resulting tree
may be incorrect
- Sequence characters
- Indels are problems when multiple
lineages have indels at the same site but the indels are not
identical (impossible to decide which came first!)
- If more than one change occurs at
a site, the second change may restore the original base
- The second occurance
of the base at the site is not phylogenetically
equal to the first
(the
second occurance is not the descendent
of the first),
although it is biochemically identical to
the first and may
be undetectable
- Theoretical Problems
- Homoplasy is common so you need to gather enough data that the true
history is supported by many characters (homoplasies tend to be unique
and supported
by only one or a few characters)
- Radiations occur so quickly that some divergences have no synapomorphies
and, thus, leave no evolutionary record
- Long-Branch Attraction
- If a phylogeny has unequal rates of evolution, such that some branches
leading to terminal taxa are long (many changes) and some very short
(few changes)
then tree construction methods will tend to place the long branches
as sister taxa, even if they are not closely related (this bias occurs
in all
methods
of tree construction)
- It is a problem that can't be solved with more data because
that usually just makes the long branches longer, which worsens
the problem
- When long branch
attraction occurs, the analysis is said to have entered
the "Felsenstein Zone", a kind of Twilight Zone
(from the TV scifi series) where
the normal rules are
turned on their heads (named after Joe Felsenstein,
who first described long branch attraction along with many
other
innovations
in phylogenetic
analysis)
- Base Composition Bias and differences in the probability of Transitions versus Tranversions
- both of these biases, if undetected, can constrain evolution and,
if the model of evolution used to score a tree does not take them
into consideration,
they may result in the acceptance of an incorrect tree
Hybridization,
Horizontal Gene Transfer, and
Gene Duplication
These three process all violate the model of
evolution behind phylogenetic tree construction, specifically the basic
tenet that says a taxon splits into daughter taxa (in the strictest sense,
this splitting is only into two daughter taxa
- This results in a bifurcating
tree in which all branching events have two descendent branches
- Trees often result from analyses of particular
data sets that have trifurcations or more branches from one Node (a branching
event) but the strict model assumes that this is a result of insufficient
data and more data would resolve all branching events into bifurcations
(not always true!)
- Evolution that can't be depicted as bifurcations
(or tri- etc. furcations) is called Reticulate Evolution
Horizontal Gene Transfer
- This is the transfer of genes between different
lineages outside of sexual reproduction (reproduction is seen as vertical
gene transfer between generations)
- Bacterial parasexual recombination is considered
HGT when the gene is transferred by transduction, transformation, or conjugation
if the recipient is an unrelated lineage (another species or another subspecies)
- Thus, a gene with a completely different ancestry
is suddenly found in a species and, if you are using that gene to understand
the lineage, you will draw the wrong conclusions
- Sequence analysis has shown that this is not
a rare event for prokaryotes and, although less common, is found in eukaryotes
as well
- Eukaryotic processes of transfer are not as
well understood but may involve eukaryotic parallels to both transduction
and transformation
- Most HGT involves environmental genes
- Housekeeping genes - those with products that function in basic cellular
processes like DNA replication, protein synthesis, etc.
- Environmental genes -
those with products that are important only in particular environments
like genes for assimilation of particular
nutrients,
genes
for disease resistance, etc.
- Housekeeping genes are optimized for interaction among themselves
as the basic cell functions are all interconnected and transfer of
these genes
disrupts the optimization (usually)
- Environmental genes are optimized for performance in particular situations
and may lose value when the environment changes but, if HGT
brings them at the right time, may be very valuable additions to the
genome
Hybridization
- When hybrids form, two lineages are merged
into one, exactly the opposite of a bifurcation
- The resulting linage may lose some of the duplicated
loci but for those loci that are not lost, the effect is the same as a
gene duplication (discussed below)
Gene Duplication
- When segments of a chromosome are duplicated
(a hybridization event is only one way for this to occur
and duplication of portions of a chromosome appear to be more common)
it complicates the
idea of ancestry because related sequences are now found at different
loci
- Orthologs -
these are two different variants at the same locus (these are what
we commonly refer to as alleles when they occur in the same species)
- Paralogs - these are two different copies of
a gene that are now at different loci due to the duplication (I don't
want to call them alleles)
- Because recombination occurs only for sequences
at the same locus, two mutations in different positions on orthologs
can
eventually be in the same sequence
- Two mutations, each on a different paralog, will
never be in the same sequence as there can be no recombination
- Thus, the evolutionary history of duplicated
genes is sundered at the time of duplication, although they continue to
reflect a common history from the time prior to the duplication
Parallelisms, Convergences
and Reversals
Although sequence data is commonly used to infer
phylogenetics, we should not let it obscure the patterns found in phenotypic
evolution that are illuminated though phylogenetic analysis and we will,
in this and the next section, investigate some of those patterns
Homoplasy is not uncommon in sequence or phenotypic
evolution and Convergence, Parallelism, and Reversals are all sources of
phenotypic homoplasy
- Convergence is the development of similar phenotypes
in response to similar environmental pressures (opportunities? - the phenotypes
are said to converge from two different ancestral phenotypes to a single
phenotype)
- Camera eyes have arisen twice and are an example
of convergent evolution
- Note that, because the convergence may involve
different parts of the bodies, that the final product, the convergent phenotypes,
can have significant differences (note the smart way that the mollusc eye
is innervated and the stupid way that the vertebrate eye is innervated)
- Parallelisms differ from Convergences
- Parallel phenotypes
are those that have arisen more than once in a phenotypic tree and
are essentially the same change
that arises in different lineages
- This means that a parallel phenotypes share very
similar developmental pathways and that the changes may be mutations to
the same genes that occurred in different lineages
- What Convergence and Parallelism share is that
each phenotype arises as an adaptation to the same environmental challenge
- What they do not share is their origins
- Example - both pandas and humans have opposable digits on their anterior
limbs but the human thumb is one of the ancestral five digits
while the panda uses an extension of a bone found in both the panda
and human
wrist
- This is a convergence as the opposition is useful for manipulation
of objects but is not a parallelism because the developmental pathways
differ
- However,
it must be said that, in many cases we do not know enough about the
genetics of complex phenotypes to separate convergences from
parallels
- Reversals
- This is the re-acquisition of a primitive character from a derived
character
- Molecular reversals are not uncommon for point mutations or for
amino acid substitutions because the number of options for the
phenotype
are very
limited
- Reversals of complex characters may not truly be reversals but
may be convergences or parallels that occur over time in the same
lineage
- The book notes the re-acquisition of lower-jaw teeth in a species
of frog
- If the genetic mechanism and phenotype
of the "reacquired" phenotype
are similar to those in the primitive condition, then it is a reversal
- If the new lower-jaw teeth differ in the genes and developmental
pathway such that the teeth are not really the same as those present
in the
ancestral phenotype, then this is a case of convergence or reversal
Last updated January 20, 2010
